




Notebooks

Categories



Cells
Premium


BioTuring
Spatial transcriptomics (ST) technology has allowed to capture of topographical gene expression profiling of tumor tissues, but single-cell resolution is potentially lost. Identifying cell identities in ST datasets from tumors or other samples remains challenging for existing cell-type deconvolution methods.
Spatial Cellular Estimator for Tumors (SpaCET) is an R package for analyzing cancer ST datasets to estimate cell lineages and intercellular interactions in the tumor microenvironment. Generally, SpaCET infers the malignant cell fraction through a gene pattern dictionary, then calibrates local cell densities and determines immune and stromal cell lineage fractions using a constrained regression model. Finally, the method can reveal putative cell-cell interactions in the tumor microenvironment.
In this notebook, we will illustrate an example workflow for cell type deconvolution and interaction analysis on breast cancer ST data from 10X Visium. The notebook is inspired by SpaCET's vignettes and modified to demonstrate how the tool works on BioTuring's platform.

BioTuring
Geneformer is a foundation transformer model pretrained on a large-scale corpus of ~30 million single cell transcriptomes to enable context-aware predictions in settings with limited data in network biology. Here, we will demonstrate a basic workflow to work with ***Geneformer*** models.
These notebooks include the instruction to:
1. Prepare input datasets
2. Finetune Geneformer model to perform specific task
3. Using finetuning models for cell classification and gene classification application

BioTuring
Advances in multi-omics have led to an explosion of multimodal datasets to address questions from basic biology to translation. While these data provide novel opportunities for discovery, they also pose management and analysis challenges, thus motivating the development of tailored computational solutions. `muon` is a Python framework for multimodal omics.
It introduces multimodal data containers as `MuData` object. The package also provides state of the art methods for multi-omics data integration. `muon` allows the analysis of both unimodal omics and multimodal omics.

BioTuring
Power analyses are considered important factors in designing high-quality experiments. However, such analyses remain a challenge in single-cell RNA-seq studies due to the presence of hierarchical structure within the data (Zimmerman et al., 2021). As cells sampled from the same individual share genetic and environmental backgrounds, these cells are more correlated than cells sampled from different individuals. Currently, most power analyses and hypothesis tests (e.g., differential expression) in scRNA-seq data treat cells as if they were independent, thus ignoring the intra-sample correlation, which could lead to incorrect inferences.
Hierarchicell (Zimmerman, K.D. and Langefeld, C.D., 2021) is an R package proposed to estimate power for testing hypotheses of differential expression in scRNA-seq data while considering the hierarchical correlation structure that exists in the data. The method offers four important categories of functions: data loading and cleaning, empirical estimation of distributions, simulating expression data, and computing type 1 error or power.
In this notebook, we will illustrate an example workflow of Hierarchicell. The notebook is inspired by Hierarchicell's vignette and modified to demonstrate how the tool works on BioTuring's platform.
Trends
BioTuring
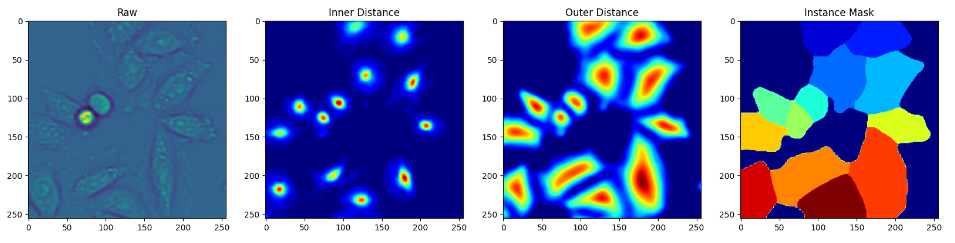
Live-cell imaging has opened an exciting window into the role cellular heterogeneity plays in dynamic, living systems. A major critical challenge for this class of experiments is the problem of image segmentation, or determining which parts of a microscope image correspond to which individual cells. Deepcell shows that deep convolutional neural networks, a supervised machine learning method, can solve this challenge for multiple cell types. The authors share their experience in designing and optimizing deep convolutional neural networks for this task and propose some design rules to achieve stable performance. The authors conclude that deep convolutional neural networks are an accurate, time-saving, applicable method for many types of cells, from bacteria to animal cells, and expand the capabilities of live-cell imaging to include multi-cell systems.
Deepcell library allows users to apply pre-existing models to imaging data as well as to develop new deep learning models for single-cell analysis. This library specializes in models for cell segmentation (whole-cell and nuclear) in 2D and 3D images as well as cell tracking in 2D time-lapse datasets. These models are applicable to data ranging from multiplexed images of tissues to dynamic live-cell imaging movies.
deepcell-tf which is written in Python using TensorFlow, is a deep learning library for single-cell analysis of biological images. It is one of several resources created by the Van Valen lab to facilitate the development and application of new deep learning methods to biology.